

Proteomica
La caratterizzazione strutturale di peptidi e proteine, così come l'identificazione di proteine non ancora ben caratterizzate dal punto di vista funzionale, costituiscono attività di rilievo nell’ambito delle biotecnologie, sia per la ricerca di base che applicata. La facility di Proteomica del CEINGE è una piattaforma di alta tecnologia che fornisce una serie di servizi analitici volti alla caratterizzazione strutturale di peptidi, proteine e loro interazioni con ligandi, basate sull'integrazione di procedure biochimiche classiche con metodologie avanzate di spettrometria di massa.
LE APPARECCHIATURE
SISTEMI ACCOPPIATI LC-MSMS
- Spettrometro di massa alta risoluzione, EXPLORIS 240 accoppiato a sistema nanoUPLC Vanquish-Neo (ThermoFisher Scientific);
- Spettrometro di massa alta risoluzione LTQ Orbitrap XL con ETD (ThermoFisher Scientific) accoppiato a sistema nanoUPLC nanoAcquity (WATERS);
- Xevo TQ-S nanoLC Triple Quadrupole con IonKey accoppiato a sistema nanoUPLC nanoAcquity (WATERS);
- Spettrometro di massa alta risoluzione 6530 Q-TOF accoppiato a sistema HPLC 1200 (Agilent Technologies);
- Spettrometro di massa 5500 Q-TRAP accoppiato a sistema HPLC ExionLC (AB Sciex).
RISONANZA PLASMONICA DI SUPERFICIE (SPR)
- Biacore X-100 (Cytiva)
I SERVIZI
Spettrometria di Massa
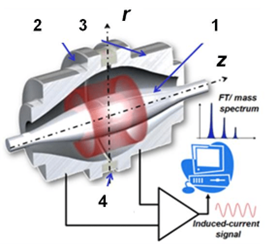
- Determinazione del peso molecolare accurato di biomolecole
Determinazione del peso molecolare accurato di proteine, peptidi, oligonucleotidi sintetici e piccole molecole (es. mataboliti) mediante analisi diretta ESI-MS e/o LC-ESI-MS.
- Analisi per quantificazione assoluta di metaboliti mediante MRM
Estrazione del metabolita di interesse da campioni biologici. Messa a punto del metodo LC-MSMS-MRM (Multiple Reaction Monitoring) su standard di riferimento per la selezione delle transizioni specifiche da monitorare. Quantificazione assoluta mediante interpolazione con retta di calibrazione costruita con quantità note di standard. Utilizzo di standard interno marcato per procedure di normalizzazione.
- Peptide mapping per analisi di modifiche post-traduzionali e varianti proteiche
Completa caratterizzazione della struttura primaria di proteine native e ricombinanti; identificazione di modifiche post-traduzionali quali modifiche epigenetiche, maturazione proteolitica e assegnazione dei ponti S-S.
- Analisi di sequenza di peptidi mediante spettrometria di massa tandem
Separazione di peptidi mediante HPLC o analisi diretta di campioni purificati e determinazione della sequenza mediante esperimenti di spettrometria di massa tandem.
Proteomica

- Separazione SDS-PAGE di campioni proteici
Frazionamento di campioni proteici mediante SDS-PAGE finalizzata alla successiva identificazione delle bande proteiche.
- Identificazione di proteine mediante LC-MS/MS
Identificazione di proteine in soluzione o frazionate mediante SDS-PAGE. Idrolisi secondo procedure shotgun (in soluzione) o in situ (per bande da gel); analisi LC-MS/MS della miscela di peptidi, identificazione delle proteine mediante ricerca in banche dati.
- Esperimenti di proteomica differenziale (tipologia “untargeted label free”) associata ad analisi LC-MS/MS per la quantificazione relativa di proteine
Estrazione di proteine da tessuti, anche FFPE e/o da cellule, quantificazione dell’estratto proteico, idrolisi con tripsina secondo procedure shotgun label free; analisi LC-MS/MS (metodi DDA, DIA) utilizzando spettrometri di massa ad alta risoluzione. Identificazione, quantificazione e analisi statistica per la determinazione delle proteine significativamente sovra- o sotto-espresse, in accordo con i Fold Changes misurati.
- Analisi “targeted” per quantificazione relativa e/o assoluta di proteine mediante MRM
Estrazione di proteine da tessuti, anche FFPE e/o da cellule, quantificazione dell’estratto proteico, idrolisi con tripsina secondo procedure shotgun. Messa a punto del metodo LC-MSMS-MRM (Multiple Reaction Monitoring) a partire da almeno due peptidi proteotipici con selezione delle transizioni specifiche. Analisi MRM su sistemi ifenati costituiti da (nano)UPLC accoppiati a spettrometri di massa a triplo quadrupolo. Quantificazione assoluta della proteina di interesse mediante interpolazione con retta di calibrazione costruita con quantità note di peptidi sintetici (standard), identici a quelli monitorati in modalità MRM. Utilizzo di standard interno marcato per procedure di normalizzazione.
Interattomica
- Analisi di interazioni tra biomolecole e ligandi mediante Surface Plasmon Resonance
Screening di binding, misura di parametri termodinamici (KD) e cinetici (Kon e Koff) dell’interazione;
- Purificazione ed identificazione di interattomi mediante esperimenti di AP-MS o IP-MS
Esperimenti in batch finalizzati alla purificazione di specifiche proteine e/o di complessi proteici utilizzando procedure di affinità (AP) oppure strategie di immunoprecipitazione (IP). Entrambi gli approcci prevedono lisi cellulare, quantificazione proteica, pre-cleaning, purificazione dei complessi e preliminare verifica della presenza della proteina esca. L’identificazione degli interattori è effettuata in accordo alle procedure già descritte.
Tutti i servizi analitici includono:
- Specifica consulenza per la definizione delle strategie più idonee al conseguimento dell’obiettivo scientifico;
- Report sui risultati e le procedure analitiche eseguite (su richiesta anche in inglese);
- Conversione dei dati in un formato adatto alla pubblicazione;
- Comunicazione scritta dei risultati e trasferimento dei dati grezzi mediante procedure certificate a tutela della loro riservatezza;
- Adeguata scontistica per analisi multiple o per numeri elevati di campioni.