

Proteomics
Structural characterization and identification of peptides and proteins, particularly those that are not yet well characterized from a functional point of view, are important activities in biotechnology for both basic and applied research. The CEINGE Proteomics and Mass Spectrometry Facility is a cutting-edge platform offering a variety of analytical services for the structural characterization of peptides and proteins, as well as their interactions with ligands. These services integrate classical biochemical procedures with advanced mass spectrometry methodologies.
EQUIPMENT
LC-MSMS COUPLED SYSTEMS
• High-resolution mass spectrometer, EXPLORIS 240 coupled with Vanquish-Neo nanoUPLC system (ThermoFisher Scientific);
• High-resolution LTQ Orbitrap XL mass spectrometer with ETD (ThermoFisher Scientific) coupled with nanoAcquity nanoUPLC system (WATERS);
• Xevo TQ-S nanoLC Triple Quadrupole with IonKey coupled to nanoAcquity nanoUPLC system (WATERS);
• 6530 Q-TOF high-resolution mass spectrometer coupled to 1200 HPLC system (Agilent Technologies);
• 5500 Q-TRAP mass spectrometer coupled with ExionLC HPLC system (AB Sciex).
SURFACE PLASMON RESONANCE (SPR)
• Biacore X-100 (Cytiva)
SERVICES
Mass Spectrometry
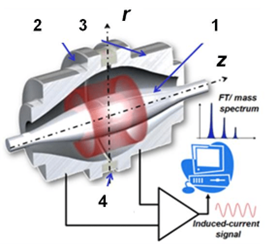
Accurate determination of the molecular weight of biomolecules
Accurate determination of the molecular weight of proteins, peptides, synthetic oligonucleotides and small molecules (e.g. metabolites) using direct ESI-MS and/or LC-ESI-MS analysis.
Analysis for the absolute quantification of metabolites using MRM
Extraction of the metabolite of interest from biological samples. Development of the LC-MSMS-MRM method using reference standards to select specific transitions to monitor. Absolute quantification by interpolation with a calibration curve constructed using known quantities of standards. Use of labelled internal standards for normalisation procedures.
Peptide mapping for the analysis of post-translational modifications and protein variants
Complete characterisation of the primary structure of native and recombinant proteins, including the identification of post-translational modifications such as epigenetic modifications, proteolytic maturation, and the assignment of S-S bridges.
Peptide sequence analysis by tandem mass spectrometry
Peptides are separated by HPLC or analysed directly from purified samples, and their sequences are determined by tandem mass spectrometry experiments.
Proteomics

SDS-PAGE separation of protein samples
Fractionation of protein samples using SDS-PAGE for subsequent identification of protein bands.
Protein identification by LC-MS/MS
Identification of proteins in solution or fractionated by SDS-PAGE. Hydrolysis according to shotgun (in solution) or in situ (for gel bands) procedures; LC-MS/MS analysis of the peptide mixture, Protein identification by database search
Differential proteomics experiments (untargeted, label-free type) are combined with LC-MS/MS analysis to quantify proteins relatively
Extraction of proteins from tissues (including FFPE) and/or cells. Quantification of the protein extract. Hydrolysis with trypsin according to label-free shotgun procedures. LC-MS/MS analysis using high-resolution mass spectrometers (DDA and DIA methods) Identification, quantification and statistical analysis to determine significantly over- or under-expressed proteins in accordance with measured fold changes.
Targeted analysis for relative and/or absolute quantification of proteins using MRM
Extraction of proteins from tissues (including FFPE) and/or cells; Quantification of the protein extract; Hydrolysis with trypsin according to shotgun procedures. Development of the LC-MSMS-MRM (multiple reaction monitoring) method, starting from at least two proteotypic peptides, with selection of specific transitions. MRM analysis on ifenate systems consisting of nanoUPLC coupled with triple quadrupole mass spectrometers. Absolute quantification of the protein of interest by interpolation with a calibration curve constructed using known quantities of synthetic peptides (standards) that are identical to those monitored in MRM mode. Use of labelled internal standards for normalisation procedures.
Interactomics
Analysis of interactions between biomolecules and ligands using surface plasmon resonance.
Binding screening and measurement of thermodynamic (K_D) and kinetic (K_(on) and K_(off)) parameters of the interaction
Purification and identification of interactomes using affinity purification mass spectrometry (AP-MS) or immunoprecipitation mass spectrometry (IP-MS) experiments
Batch experiments aimed at purifying specific proteins and/or protein complexes using affinity (AP) procedures or immunoprecipitation (IP) strategies. Both approaches involve cell lysis, protein quantification, pre-cleaning, complex purification, and preliminary verification of the bait protein's presence. Identification of interactors is carried out in accordance with the previously described procedures.
All analytical services include:
- Specific consulting to define the most suitable strategies to achieve the scientific objective;
- Reporting of results and analytical procedures performed (also available in English upon request);
- Data conversion into a format suitable for publication;
- Written communication of results and transfer of raw data using certified procedures to protect confidentiality;
- Appropriate discounts for multiple analyses or large numbers of samples.